
Pentacle
Advanced Alignment-Independent 3D QSAR
Pentacle is an advanced software tool for obtaining alignment-independent 3D quantitative structure-activity relationships. The program starts from a set of structures, computing highly relevant 3D maps of interaction energies between the molecule and chemical probes (GRID based Molecular Interaction Fields, or MIFs). Pentacle automatically encodes these maps into GRID Independent Descriptors (GRIND and GRIND2 descriptors), which are independent of the alignment of the series, thus bypassing one of the most time-consuming aspects of traditional 3D QSAR.
Starting from 3D structures of an active series of compounds, Pentacle is able to build a model within a couple of minutes, and guide the user step-by-step through extracting the most useful information
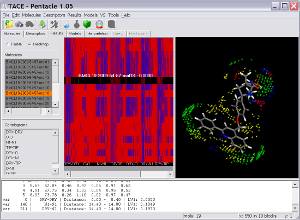
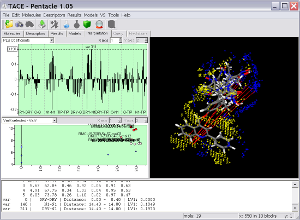
Key Features
- Automatically calculate GRID-based GRIND & GRIND2 descriptors
- Create and explore models interactively using advanced multivariate analyses, linked to the 3D Molecular Interaction Fields of the series
- Automatically align structures to aid interpretation using the new CLACC algorithm
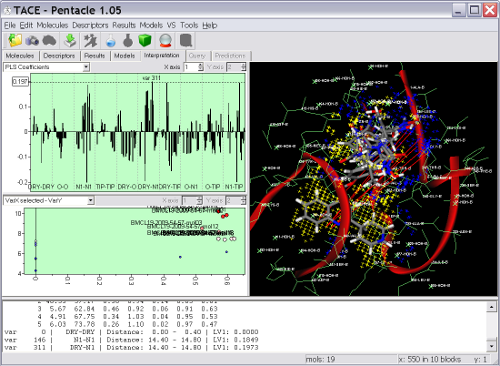
Pentacle includes new MIF sampling and encoding algorithms improving on those previously implemented for the Almond GRIND 3D QSAR program. The example shows several TACE inhibitors and the Pentacle 3D QSAR model. One of the important contributions to the activity of the series is a polar interaction (illustrated by the red vectors between the yellow hydrophobic field nodes to the blue hydrogen bond donor field nodes) with Thr347. Pentacle is able to import the compounds, calculate the descriptors, build a model, and help you interpret the model in just couple of minutes.
Key References
- Pastor, M.; McLay, I.; Pickett, S.; Clementi, S. Grid-Independent descriptors (GRIND): a novel class of alignment-independent three-dimensional molecular descriptors. Med. Chem. 2000, 43 (17), 3233-3243.
- Duran, A.; Comesaña G, Pastor, M. Development and validation of AMANDA, a new algorithm for selecting highly relevant regions in molecular interaction fields. J. Chem. Inf. Model. 2008, 48 (9), 1813-1823.
- Duran, A.; Zamora, I; Pastor, M. Suitability of GRIND-Based Principal Properties for the Description of Molecular Similarity and Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2009, 49 (9), 2129-2138.