
Mass-MetaSite
High-Throughput MetID
Mass-MetaSite is an established approach for the automatic identification of metabolites for small molecules and peptides using Liquid Chromatography - Mass Spectrometry, UV, fluorescencem, and radio-chromatogram data, reducing manual analysis from several hours to only a few minutes per compound [1]. The program is able to assign chemical structures to each automatically detected chromatographic peak based on the MS and MS/MS fragmentation pattern of the substrates and metabolites. It can process from multiple vendors (Agilent, Bruker, Sciex, Thermo, and Waters) and it is also available to analyze data from different acquisition modes (DDA, MSE, HDMSE, AIF, AF, Broad band, SWATH, Sonar, etc.)
In the cases where the data cannot be used to propose a single structure for the chromatographic peaks found, the system also introduces the Site of Metabolism (SoM) prediction from MetaSite computation (the leader in the metabolism prediction market), that ranks the multiple structural options. Moreover, the user has access to the visual analysis of the enzyme-metabolite interaction CYPs, FMO and AOX proteins, and it can even propose structural modification to overcome the metabolic liability.
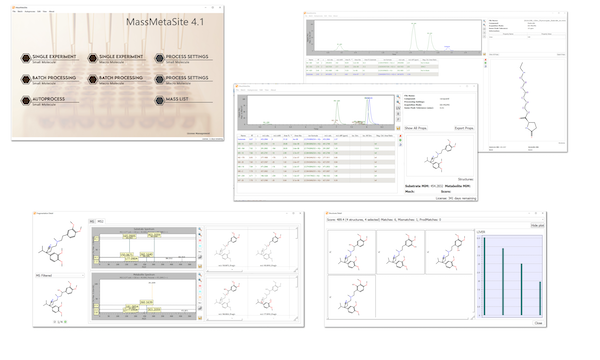
The auto-process and the batch processor enable the use of the software for automatic process in ADME workflows like GSH, Met ID, Soft Spot and much more.
A revolution in MetID for drug discovery
- Agilent Q-Tof(*.d): AutoMS and full scan at multiple energies of collision.
- Waters (*.raw, UNIFI): MSe, HDMSe, DDA, MSMS and MRM.
- Thermo-Fisher (*.RAW): Ion-Trap and Orbitrap Data Dependent Scan, Exactive, Q-Exactive.
- ABSCiex *.wiff file format, single wiff file containing multiple samples supported.
- Bruker (*.d): QTof and TIMS-TOF data dependent scan.
Reading of the most common file formats:
Features in MassMetasite
- Automatic peak detection
- Structure elucidation
- Comparison with parent
- Metabolite specific fragmentation
- Batch processor:
- Auto-processing of experiment files
- Multiple process batches
- New tool to create WebMetabase Protocol Instances/Batches on sample list import time
- Setting/mode to process data for calibration curves
- Exporting to excel report
- Markush handling depiction
- Modes of operation:
- Standard
- GSH: Neutral lost and fragment ions
- Cyanide
- Peptide (up to 4500 amu)
- Signal Process:
- Mass Spectrometry, UV, Fluorescence and Radio files
- Direct connection to design tools:
- MetaDesign module available, 32D, MetaSite, Metabolite generation, Automatic validation
- Isotope labeling:
- Stable GSH and cyanide isotope labeling
- Radio isotope labeling
- Enhanced peak quality analysis:
- Area compared to the Blank
- Area compared to the Substrate
- Isotope similarity score
- Improved connection to UNIFI
Small molecules:
- Handle oligonucleotides, macro-cyclic peptides (MCP), and other molecules classes (up to 50.000 amu).
- Automatic peak detection
- Structure elucidation:
- Comparison with parent
- Metabolite specific fragmentation
- Batch processor:
- Multiple process batches
- Tool to create WebMetabase Protocol Instances/Batches on sample list import time
- Visualization in monomers and/or atomic notation
- Signal process:
- Mass Spectrometry, UV, Fluorescence and Radio files
- Isotope labeling:
- Radio isotope labeling
- Enhanced peak quality analysis:
- Area compared to the Blank
- Area compared to the Substrate
- Isotope similarity score
- Improved connection to UNIFI
Macromolecules:
MassMetaSite 4.6.0 Release Notes
- Tagging of metabolie names based on custom mass shifts
- Custom biotransformations based on formula changes
- Automatic update of metabolite formulas based on selected structures in detail view
- Bruker tsf files supported
- Agilent SLIM files supported
General MassMetaSite update:
MassMetaSite 4.5.1 Release Notes
- MS file converter added to the installation package
- Improved management of batches and settings
- Improved management of WebMetabase Protocols
- Improves reaction management in Settings
- Increased speed of peak detection
- Increased speed during structure loading
General MassMetaSite update:
MassMetaSite Macro molecule update:
MassMetaSite 4.5.0 Release Notes
- Sciex Multiple Reaction Monitoring (MRM)
- Improved management of batches and settings
- Added special reactions: Dimethylation (N-terminus), O-dealkylation linear
- Improved fragmentation algorithm
General MassMetaSite update:
MassMetaSite Macro molecule update:
MassMetaSite 4.4.2 Release Notes
- Custom lock mass m/z
- Increased number of decimals for internal standard m/z
- Special mode to fragment β-γ bonds only
- Improved most abundant mass detection algorithm
General MassMetaSite update:
MassMetaSite Macro molecule update:
MassMetaSite 4.3.0 Release Notes
- Acquisition modes:
- Waters QQQ
- Wiff file with multiple samples
- SMS file size reduction
- New sulphoximine reactions: Sulphoximine Oxidation, Sulphoximine Deamination and Sulphoximine N-Dealkylation
- MassMetaSite Macro molecule update:
- Oligonucleotides, macro-cyclic peptides (MCP), and other molecules classes (up to 50.000 amu).
- Customized linear visualization for larger molecules with more than 100 monomers
- Average Mass calculation
- Charge deconvolution
- Fine tuning of reaction metabolism: i.e. metabolize from termini only. Metabolize by intra monomer reaction groups
- Added new peptide-specific intra monomer reactions
- Improve elucidation by fragmenting metabolites (metmatches)
General MassMetaSite update:
Please login to see an overview of the Mass-MetaSite capabilities.
References
- [1] Enhanced
metabolite identification with MS(E) and a semi-automated software
for structural elucidation.
Bonn B, Leandersson C, Fontaine F, Zamora I. Rapid Commun Mass Spectrom. 2010 Nov 15;24(21):3127-38. - [2] High-throughput, fully automated, specific MetID. A revolution for Drug Discovery.
Zamora I, Fontaine F, Serra B, Plasencia G, Drug Discovery Today: Technologies 2012, in press. - [3] Metabolism of JWH-015, JWH-098, JWH-251, and JWH-307 in silico and in vitro:
a pilot study for the detection of unknown synthetic cannabinoids metabolites.
Strano-Rossi S1, Anzillotti L, Dragoni S, Pellegrino RM, Goracci L, Pascali VL, Cruciani G. Analytical and Bioanalytical Chemistry.June 2014;406(15):3621-3636. - [4] High-throughput, computer assisted, specific MetID. A revolution for drug discovery.
Ismael Zamora, Fabien Fontaine, Blanca Serra, Guillem Plasencia. Drug Discovery Today: Technologies, 2013 Spring Issue;10(1):e199–e205. - [5] Software automation tools for increased throughput metabolic soft-spot identification in early drug discovery.
Veronica Zelesky, Richard Schneider1, John Janiszewski1, Ismael Zamora, James Ferguson & Matthew Troutman. Bioanalysis, 5(10):1165-1179. - [6] Update on hydrocodone metabolites in rats and dogs aided with a semi-automatic software for metabolite
identification Mass-MetaSite.
Austin C. Li, James P. Chovan, Erya Yu, and Ismael Zamora. Xenobiotica, April 2013;43(4):390-398 - [7] Enhanced metabolite identification with MSE and a semi-automated software for structural elucidation.
Britta Bonn1, Carina Leandersson, Fabien Fontaine, Ismael Zamora. Rapid Communications in Mass Spectrometry , 15 November 2010;24(21):3127–3138. - [8] Post-acquisition analysis of untargeted accurate mass quadrupole time-of-flight MS(E)
data for multiple collision-induced neutral losses and fragment ions of glutathione conjugates.
Brink A, Fontaine F, Marschmann M, Steinhuber B, Cece EN, Zamora I, Pähler A. Rapid Commun Mass Spectrom. 2014 Dec 30;28(24):2695-703