
GRID 2021
Structure-Based Design & Fragment Replacement
GRID is a computational procedure for determining energetically favourable binding sites on molecules of known structure, known as Molecular Interaction Fields (MIFs).
The best-known application of GRID in Structure-Based Drug Design [ Nature 363, 418-423 (1993) ] shows how potent inhibitors based on the crystal structure of influence virus sialidase were designed, leading to the approved drug RELENZA®. GRID MIFs have been used for a wide range of applications including ADME prediction (VolSurf+), Site of Metabolism Prediction (MetaSite), Ligand-Based and Structure-Based Design (FLAP), Pharmacophore elucidation (FLAP), water network prediction and scoring (WaterFLAP), and 3D-QSAR (FLAP)
GRID 2021 is a new interface to the classic program, aimed primarily at Structure-Based Design. Users can explore binding sites using the classical GRID MIFs which include 74 different chemical types, and can additionally use the new molecular probe to generate MIFs for fragments of interest. A 3D sketcher is included so that users can visualise potential modifications to bound ligands, and Designer mode helps answer the question "which chemical moiety would fit best here?".
Key GRID features:
- Calculate MIFs using 74 different chemical types
- Calculate custom MIFs for a fragment of interest
- 3D sketcher to test ideas
- Designer mode to retrieve optimal R-Groups
- Generate single chemical mutations in a ligand to help improve binding
- Import WaterFLAP results to guide design
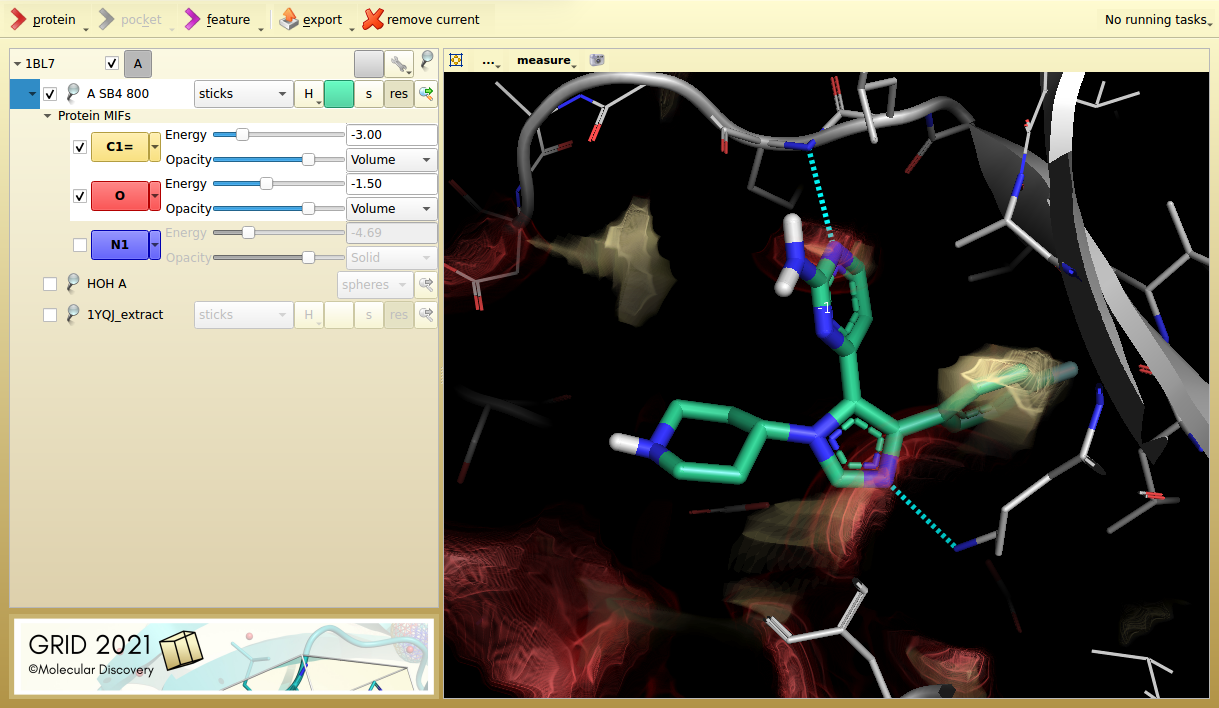
Figure 1. p38 MAP kinase inhibitor SB4 in its target protein (1BL7). The aromatic carbon probe (C1=, yellow) and the carbonyl acceptor probe (O, red), highlight key interactions.
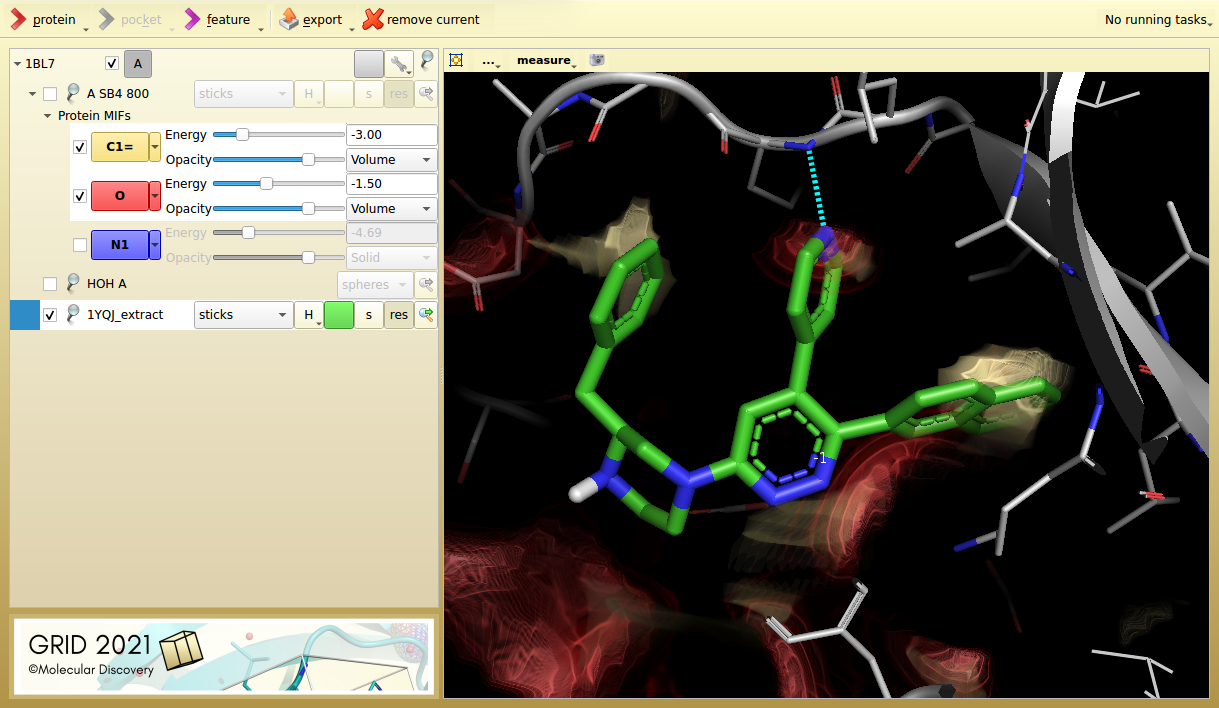
Figure 2. p38 MAP kinase inhibitor 6NP from its aligned target protein (1YQJ). The inhibitor hits an additional lipophilic hotspot and is seven times more potent (IC50 7.3 nM).
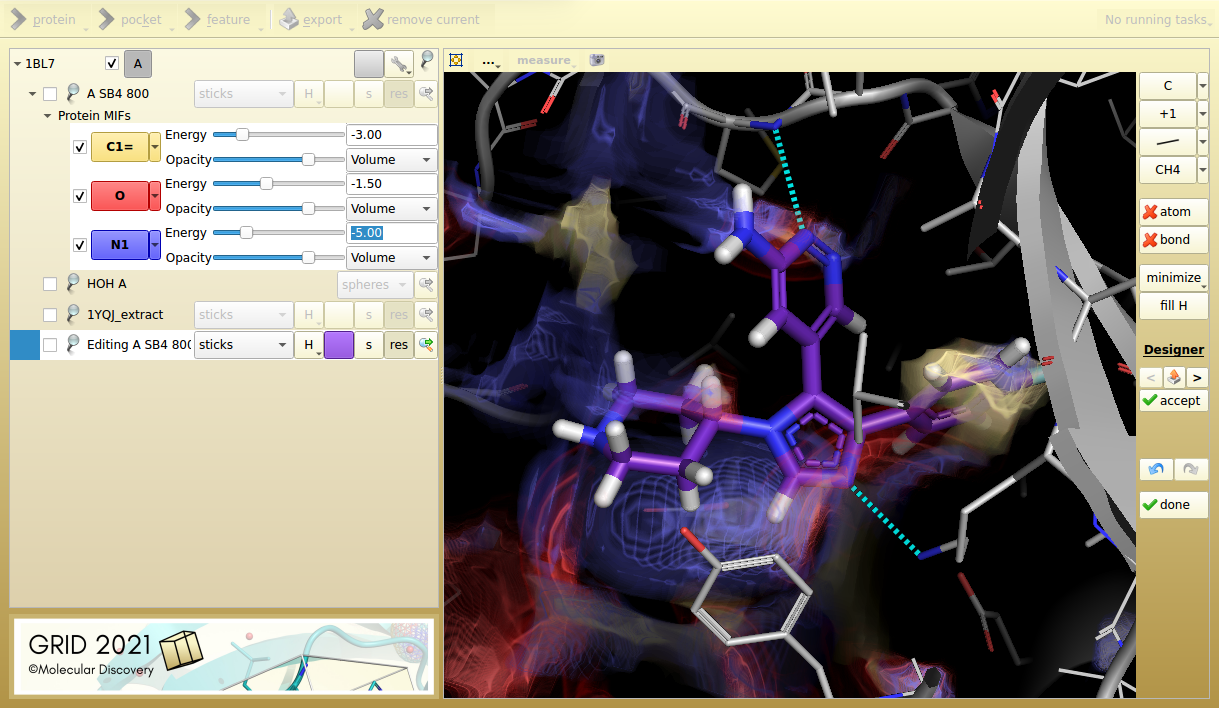
Figure 3. Designer mode shows a potential SB4 hinge-binder fragment replacement, making similar interactions.
References
-
Peter J. Goodford
A computational procedure for determining energetically favorable binding sites on biologically important macromolecules
J. Med. Chem., 1985, 28 (7), pp 849-857 -
Rebecca C. Wade, Kevin J. Clark, Peter J. Goodford
Further development of hydrogen bond functions for use in determining energetically favorable binding sites on molecules of known structure. 1. Ligand probe groups with the ability to form two hydrogen bonds
J. Med. Chem., 1993, 36 (1), pp 140-147 -
Rebecca C. Wade, Peter J. Goodford
Further development of hydrogen bond functions for use in determining energetically favorable binding sites on molecules of known structure. 2. Ligand probe groups with the ability to form more than two hydrogen bonds
J. Med. Chem., 1993, 36 (1), pp 148-156 - Emanuele Carosati, Simone Sciabola, and Gabriele Cruciani
Hydrogen Bonding Interactions of Covalently Bonded Fluorine Atoms: From Crystallographic Data to a New Angular Function in the GRID Force Field
J. Med. Chem., 2004, 47 (21), pp 5114–5125 -
Artese A, Cross S, Costa G, Distinto S, Parrotta L, Alcaro S, Ortuso F, Cruciani G.
Molecular interaction fields in drug discovery: recent advances and future perspectives.
WIREs Comput Mol Sci 2013, 3:594-613. -
Sara Tortorella, Emanuele Carosati, Giulia Sorbi, Giovanni Bocci, Simon Cross, Gabriele Cruciani, Loriano Storchi
Combining machine learning and quantum mechanics yields more chemically aware molecular descriptors for medicinal chemistry applications
J. Comp. Chem., 2021